Quick guide
Here we show a quick guide of our code and some of the plots and anslysis you can get from it.
Memb2D
This quick tutorial will include computation of
Membrane Thickness
2D order paramaters
Packing defects
Voronoi APL
Import the class, and mdanalysis
import MDAnalysis as mda
from twodanalysis import Memb2D
All this analysis have in common the definition of a class that must be done with a universe
tpr = "membrane.tpr" # Replace this with you own membrane tpr or gro file
xtc = "membrane.xtc" # Replace this with you xtc fila
universe = mda.Universe(tpr,xtc) # Define a universe with the trajectories
membrane = Memb2D(universe, # load universe
verbose = False, # Does not print intial information
add_radii = True) # Add radii (used in packing defects)
Membrane Thickness
This code requires the number of bins, the edges, and the time interval that you want to use. Other options are also available, check the documentation for mor information.
mat_thi, edges = membrane.thickness(50, # nbins
start = 61, # Initial frame
final = 110, # Final Frame
step = 1, # Frames to skip
)
The output is a matrix \(nbins\times nbins\) and the edges in the form \([xmin,xmax,ymin,ymax]\).
We can visualize with plt.imshow
import matplotlib.pyplot as plt plt.imshow(mat_thi, extent=edges, cmap="Spectral") plt.xlabel("x $\AA$") plt.ylabel("y $\AA$") plt.title("Membrane thichness from frames 61-110") cbar = plt.colorbar() cbar.set_label('Thickness $\AA$')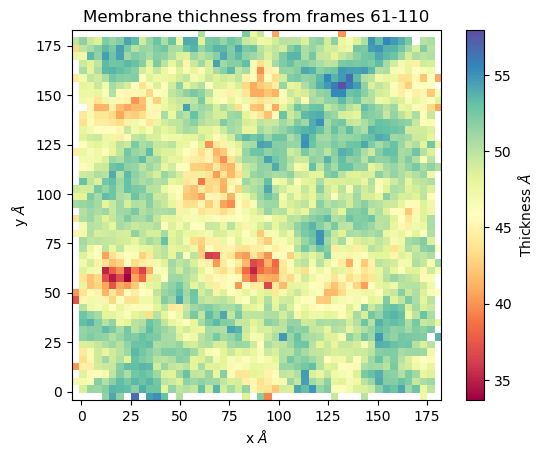
Membrane order parameters
The computation of order parameters is as easy as the computation of thickness. In this case you can also choose which layer the analysis will run (top, bot, both). Follows an example of running order parameters
scd_top, edges = membrane.thickness( "top", # top layer
50, # nbins
start = 61, # Initial frame
final = 110, # Final Frame
step = 1, # Frames to skip
)
scd_bot, edges = membrane.thickness( "bot", # bot layer
50, # nbins
start = 61, # Initial frame
final = 110, # Final Frame
step = 1, # Frames to skip
)
Now we can plot the results
from mpl_toolkits.axes_grid1 import make_axes_locatable # Plot fig, ax = plt.subplots(1,2, sharex = True, sharey = True) first = ax[0].imshow(scd_top, extent=edges, cmap="Spectral") ax[0].set_xlabel("x $\AA$") ax[0].set_ylabel("y $\AA$") ax[0].set_title("Top layer") divider1 = make_axes_locatable(ax[0]) cax1 = divider1.append_axes("right", size="5%", pad=0.05) cbar = fig.colorbar(first, cax = cax1) # Point to a low ordered region ax[0].add_patch(patches.Rectangle((48, 98), 20,20, linewidth = 1, edgecolor = "black", facecolor = "none")) # High ordered region ax[0].add_patch(patches.Rectangle((90, 120), 20,20, linewidth = 1, edgecolor = "black", facecolor = "none")) second = ax[1].imshow(scd_bot, extent=edges, cmap="Spectral") ax[1].set_xlabel("x $\AA$") ax[1].set_title("Bot layer") divider2 = make_axes_locatable(ax[1]) cax2 = divider2.append_axes("right", size="5%", pad=0.05) cbar = fig.colorbar(second, cax = cax2) cbar.set_label('|SCD| $\AA$')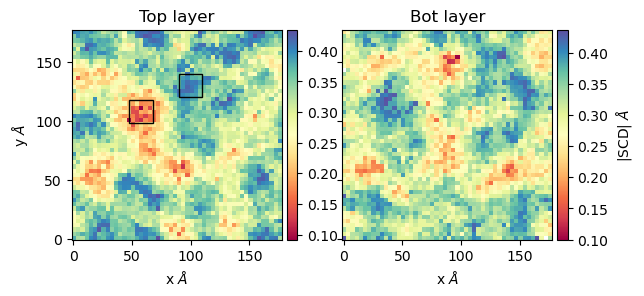
Here we highligted regions where the order parameters are low (red region) and high (blue region). From this region the lipids looks as follows

Packing defects
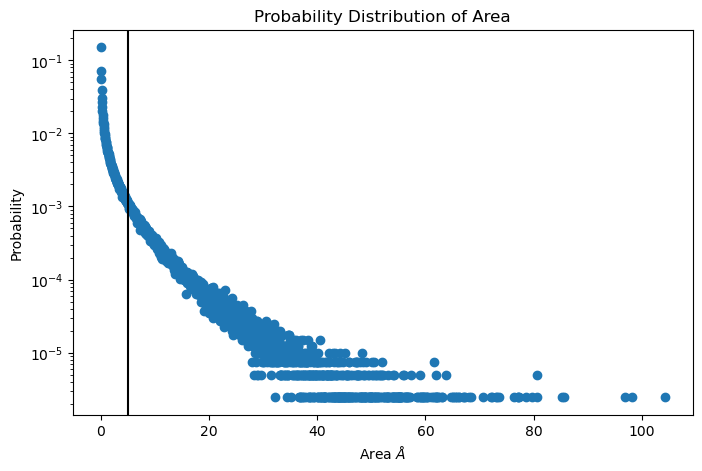
Packing defects is metric to evaluate the exposure of the hydrophobic core. It changes with membrane composition and also when proteins interact with the membrane. The computation of packing defects with packmemb implies extracting pdb files from the trajectories and then procesing them, which is time comsuming. Here we present an easy way to compute packing defects by only providing the trajectory and the topology file. Also, our code outperforms packmemb, doing the computations faster.
The packing defects code is the following:
# Compute deffects for the first frame
defects, defects_dict = membrane.packing_defects(layer = "top", # layer to compute packing defects
edges=[10,170,10,170], # edges for output
nbins = 400, # number of bins
)
# Plot defects
%matplotlib inline
plt.imshow(defects, cmap = "viridis", extent = defects_dict["edges"])
plt.xlabel("x $[\AA]$")
plt.ylabel("y $[\AA]$")
plt.show()

For various frames to get statistics
data_df, numpy_sizes = membrane.packing_defects_stats(nbins = 400,
layer = "top",
periodic = True,
start = 0,
final = -1,
step=1)
Area perlipid
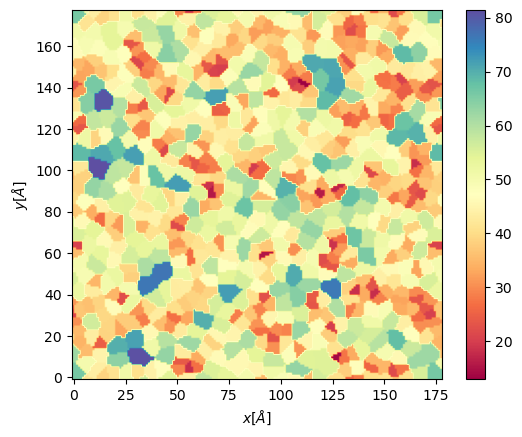
We include the posibility of get Voronoi APL. For one frame can be obtained as follows:
voronoi_dict = membrane.voronoi_apl(layer = "top")
This return a dictionary that contains the areas per each lipid in the top bilayer
We can further map this voronoi to a twod grid and plot it
xmin = membrane.v_min
xmax = membrane.v_max
ymin = membrane.v_min
ymax = membrane.v_max
apl, edges = membrane.map_voronoi(voronoi_dict["points"], voronoi_dict["areas"], 180, [xmin, xmax, ymin, ymax])
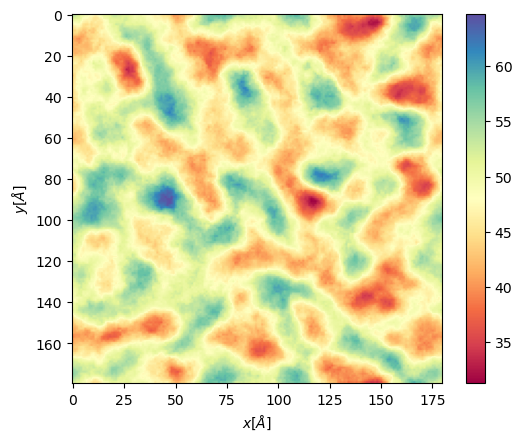
plt.imshow(apl, extent = edges, cmap = "Spectral")
plt.xlabel("$x [\AA]$")
plt.ylabel("$y [\AA]$")
plt.colorbar()
For multiples frames:
resu, edges = membrane.grid_apl(layer = "top", start = 10, final = 100, step = 1, lipid_list = None)
plt.imshow(resu, extent = edges, cmap = "Spectral")
plt.xlabel("$x [\AA]$")
plt.ylabel("$y [\AA]$")
plt.colorbar()